Zuvog M 0.2 mg Tablet


1.0 Generic Name
Voglibose and Metformin Hydrochloride (Prolonged-release) Tablets
2.0 Qualitative and quantitative composition
Each uncoated bilayered tablet contains:
Voglibose IP………………………0.2 mg/ 0.3 mg
Metformin Hydrochloride IP……...500 mg
(As Prolonged-release form)
Excipients……………………………. q.s.
3.0 Dosage form and strength
Dosage Form: Tablets.
Dosage Strength: Voglibose 0.2 mg/ 0.3 mg and Metformin Hydrochloride 500 mg
(Prolonged-release)
4.0 Clinical particulars
4.1 Therapeutic Indication
As 2nd line treatment of type II Diabetes mellitus when diet, exercise and voglibose or metformin do not result in adequate glycemic control individually.
4.2 Posology and method of administration
Dosage should be individualised on the basis of both effectiveness and tolerance.
The initial recommended dose is one tablet of ZUVOG M 0.2 three times daily just before each meal.
In case of inadequate effect, the dose may be increased up to 1 tablet of ZUVOG M 0.3, three times daily just before each meal under close observation of the course of the disease.
4.3 Contraindications
- Renal disease or renal dysfunction (e.g., as suggested by serum creatinine levels 1.5 mg/dL [males], 1.4 mg/dL [females] or abnormal creatinine clearance), which may also result from conditions such as cardiovascular collapse (shock), acute myocardial infarction, and septicaemia.
- Hypersensitivity to metformin, voglibose or any of the components of this product.
- Acute or chronic metabolic acidosis, including diabetic ketoacidosis, with or without coma. Diabetic ketoacidosis should be treated with insulin.
- Severe infection, before and after surgery, serious trauma.
- Gastrointestinal obstruction or predisposed to it.
Patients undergoing radiologic studies involving intravascular administration of iodinated contrast materials should temporarily discontinue taking this combination, because use may result in acute alteration of renal function.
4.4 Special warnings and precautions for use
Loss of Control of Blood Glucose
When a patient stabilised on any diabetic regimen is exposed to stress such as fever, trauma, infection, or surgery, a temporary loss of glycaemic control may occur. At such times, it may be necessary to withhold oral antidiabetic agents and temporarily administer insulin. This combinationmay be reinstituted after the acute episode is resolved.
Hypoxic States
Cardiovascular collapse (shock) from whatever cause, acute congestive heart failure, acute myocardial infarction and other conditions characterised by hypoxaemia have been associated with lactic acidosis and may also cause prerenalazotaemia. When such events occur in patients on ZUVOG M, the drug should be promptly discontinued.
Surgical Procedures
ZUVOG M should be temporarily suspended for any surgical procedure (except minor procedures not associated with restricted intake of food and fluids) and should not be restarted until the patient’s oral intake has resumed and renal function has been evaluated as normal.
Alcohol Intake
Alcohol is known to potentiate the effect of metformin on lactate metabolism. Patients, therefore, should be warned against excessive alcohol intake, acute or chronic, while receiving ZUVOG M.
Change in Clinical Status of Patients with Previously Controlled Type 2 Diabetes
A patient with type 2 diabetes with previously well controlled type 2 diabetes who develops laboratory abnormalities or clinical illness (especially vague and poorly defined illness) should be evaluated promptly for evidence of ketoacidosis or lactic acidosis. Evaluation should include serum electrolytes and ketones, blood glucose and, if indicated, blood pH, lactate, pyruvate, and metformin levels. If acidosis of either form occurs, ZUVOG M must be stopped immediately and other appropriate corrective measures initiated.
Hypoglycaemia
Hypoglycaemia does not occur in patients receiving ZUVOG M alone under usual circumstances of use, but could occur when caloric intake is deficient, when strenuous exercise is not compensated by caloric supplementation, or during concomitant use with other glucose-lowering agents (such as sulphonylureas and insulin) or ethanol. Elderly, debilitated, or malnourished patients and those with adrenal or pituitary insufficiency or alcohol intoxication are particularly susceptible to hypoglycaemic effects. Hypoglycaemia may be difficult to recognize in the elderly, and in people who are taking beta-adrenergic blocking drugs. Patients should be instructed and explained to recognize hypoglycaemic symptoms and its management.
Vitamin B12
In controlled clinical trials of metformin of 29 weeks duration, a decrease to subnormal levels of previously normal serum vitamin B12 levels, without clinical manifestations, was observed in approximately 7% of patients. Such decrease, possibly due to interference with B12 absorption from the B12 intrinsic factor complex, however is very rarely associated with anaemia and appears to be rapidly reversible with discontinuation of metformin or vitamin B12 supplementation. Measurement of haematologic parameters on an annual basis is advised in patients on metformin and any apparent abnormalities should be appropriately investigated and managed.
Certain individuals (those with inadequate vitamin B12 or calcium intake or absorption) appear to be predisposed to developing subnormal vitamin B12 levels. In these patients, routine serum vitamin B12 measurements at 2- or 3- year intervals may be useful.
Macrovascular Outcomes
There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with voglibose, metformin or any other anti-diabetic drug.
Lactic Acidosis
Lactic acidosis is a rare, but serious, metabolic complication that can occur due to metformin accumulation during treatment with metformin; when it occurs, it is fatal in approximately 50% of cases. Lactic acidosis may also occur in association with a number of pathophysiologic conditions, including diabetes mellitus, and whenever there is significant tissue hypo perfusion and hypoxaemia. Lactic acidosis is characterised by elevated blood lactate levels (>5 mmol/L), decreased blood pH, electrolyte disturbances with an increased anion gap, and an increased lactate/pyruvate ratio. When metformin is implicated as the cause of lactic acidosis, metformin plasma levels >5 μg/mL are generally found.
The reported incidence of lactic acidosis in patients receiving metformin hydrochloride is very low. Patients with congestive heart failure requiring pharmacologic management, in particular those with unstable or acute congestive heart failure who are at risk of hypo perfusion and hypoxaemia, are at increased risk of lactic acidosis. The risk of lactic acidosis increases with the degree of renal dysfunction and the patient’s age. The risk of lactic acidosis may, therefore, be significantly decreased by regular monitoring of renal function in patients taking metformin and by use of the minimum effective dose of metformin. In particular, treatment of the elderly should be accompanied by careful monitoring of renal function. Metformin treatment should not be initiated in patients ≥80 years of age unless measurement of creatinine clearance demonstrates that renal function is not reduced, as these patients are more susceptible to developing lactic acidosis. In addition, metformin should be promptly withheld in the presence of any condition associated with hypoxaemia, dehydration, or sepsis. Because impaired hepatic function may significantly limit the ability to clear lactate, metformin should generally be avoided in patients with clinical or laboratory evidence of hepatic disease. Patients should be cautioned against excessive alcohol intake, either acute or chronic, when taking metformin, since alcohol potentiates the effects of metformin hydrochloride on lactate metabolism. In addition, metformin should be temporarily discontinued prior to any intravascular radiocontrast study and for any surgical procedure.
The onset of lactic acidosis often is subtle, and accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, increasing somnolence, and nonspecific abdominal distress. There may be associated hypothermia, hypotension, and resistant bradyarrhythmias with more marked acidosis. The patient and the patient’s physician must be aware of the possible importance of such symptoms and the patient should be instructed to notify the physician immediately if they occur. Metformin should be withdrawn until the situation is clarified. Serum electrolytes, ketones, blood glucose, and if indicated, blood pH, lactate levels, and even blood metformin levels may be useful. Once a patient is stabilised on any dose level of metformin, gastrointestinal symptoms, which are common during initiation of therapy, are unlikely to be drug related. Later occurrence of gastrointestinal symptoms could be due to lactic acidosis or other serious disease.
Levels of fasting venous plasma lactate above the upper limit of normal but less than 5 mmol/L in patients taking metformin do not necessarily indicate impending lactic acidosis and may be explainable by other mechanisms, such as poorly controlled diabetes or obesity, vigorous physical activity, or technical problems in sample handling.
Lactic acidosis should be suspected in any diabetes patient with metabolic acidosis lacking evidence of ketoacidosis (ketonuria and ketonemia).
Lactic acidosis is a medical emergency that must be treated in a hospital setting. In a patient with lactic acidosis who is taking metformin, the drug should be discontinued immediately and general supportive measures promptly instituted. Because metformin hydrochloride is dialyzable (with a clearance of up to 170 mL/min under good hemodynamic conditions), prompt haemodialysis is recommended to correct the acidosis and remove the accumulated metformin. Such management often results in prompt reversal of symptoms and recovery.
4.5 Drugs interactions
Cationic Drugs
Certain medications used concomitantly with metformin may increase the risk of lactic acidosis. Cationic drugs that are eliminated by renal tubular secretions (e.g: amiloride, cimetidine, digoxin, morphine, procainamide, quinidine, quinine, ranitidine, triamterene, trimethoprim or vancomycin) may decrease metformin elimination by competing for common renal tubular transport systems. Hence, careful patient monitoring and dose adjustment of ZUVOG M and/or interfering drug is recommended in patients who are taking cationic medications that are excreted via the proximal renal tubular secretory system.
Furosemide
A single-dose, metformin-furosemide drug interaction study in healthy subjects demonstrated that pharmacokinetic parameters of both compounds were affected by co-administration. Furosemide increased the metformin plasma and blood Cmax by 22% and blood AUC by 15%, without any significant change in metformin renal clearance. When administered with metformin, the Cmax and AUC of furosemide were 31% and 12% smaller, respectively, than when administered alone, and the terminal half-life was decreased by 32%, without any significant change in furosemide renal clearance. No information is available about the interaction of metformin and furosemide when co-administered chronically.
Nifedipine
Nifedipine appears to enhance the absorption of metformin, it increases plasma metformin Cmax and AUC by 20% and 9% respectively and increases the amount of metformin excreted in the urine. Metformin has minimal effects on nifedipine.
Danazol
If the use of this active substance cannot be avoided, warn the patients and emphasise the importance of urine and blood glucose monitoring. It may be necessary to adjust the dose of voglibose and metformin during and after treatment with danazol.
Salicylates
If salicylates are administered or discontinued in patients receiving oral antidiabetic agents, patients should be monitored for hypoglycaemia or loss of blood glucose control.
Thiazide
Interactions between thiazide diuretics and oral antidiabetic agent’s decreases insulin sensitivity thereby leading to glucose intolerance and hyperglycaemia, thus leading to a loss of diabetic control. Hence diabetic patients should be monitored closely.
Other
Concomitant administration of angiotensin enzyme inhibitors (captopril, enalapril), other antidiabetic drugs (insulin, acarbose) beta-blockers, fluconazole, histamine (H2) receptor antagonist, monoamine oxidase inhibitors (MAOIs), sulphonamides and non-steroidal anti-inflammatory agents increase sensitivity to insulin and potentiation of blood glucose lowering effect and thus, in some instances, hypoglycaemia may occur . Certain drugs tend to produce hyperglycaemia and may lead to loss of glycaemic control. These drugs include corticosteroids, phenothiazines, thyroid products, estrogens, oral contraceptives, phenytoin, nicotinic acid, sympathomimetics, calcium channel blocking drugs, quinolones and isoniazid. Patients receiving these drugs should be closely monitored for loss of diabetic control when therapy is instituted or discontinued. Dosage of the oral antidiabetic agents may need to be reduced.
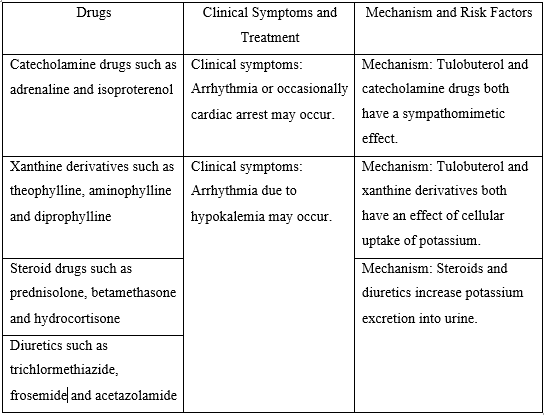
Drugs Enhancing (beta-blockers, salicylic acid preparations, MAOs, fibrate derivatives) or Diminishing (epinephrine, adrenocortical hormone, thyroid hormone etc.) the Hypoglycaemic Action of Anti-Diabetic Drugs
When ZUVOG M is administered concomitantly with drugs that enhance or diminish the hypoglycaemic action of anti-diabetic drugs, caution should be taken as this might additionally delay the action of voglibose on the absorption of carbohydrates.
Warfarin
Metformin and voglibose do not affect the pharmacokinetics of warfarin, hence ZUVOG M can be safely administered along with warfarin.
4.6 Use in special populations
Pregnant Women
Recent information suggests that abnormal blood glucose levels during pregnancy are associated with the higher incidence of congenital abnormalities. There are no adequate and well-controlled studies in pregnant women with metformin. Metformin was not teratogenic in rats and rabbits at doses up to 600 mg/kg/day. The safety and effectiveness of voglibose in pregnant women has not been established. Animal studies have shown that voglibose is transferred to the foetus. ZUVOG M should be used during pregnancy only if the potential benefit justifies the potential risk to the foetus. Most experts suggest insulin be used to maintain the blood glucose levels as close to normal as possible.
Lactating Women
Studies in lactating rats show that metformin is excreted into milk and reaches levels comparable to those in plasma. Similar studies have not been conducted on nursing mothers. Animal studies have shown a suppressive action of voglibose on body weight increase in newborns, mainly due to suppression of milk production resulting from inhibition of carbohydrate absorption in mother animals. Nursing should be discontinued if ZUVOG M has to be administered. If the use of this combination is discontinued, and if the diet alone is inadequate for controlling blood glucose, insulin therapy should be considered.
Paediatric Patients
The safety and effectiveness of metformin for the treatment of type 2 diabetes have been established in paediatric patients ages 10 to 16 years (studies have not been conducted in paediatric patients below the age of 10 years). The safety and effectiveness of voglibose in children has not been established. Hence ZUVOG M should not be considered in this population.
Geriatric
Metformin is known to be excreted through the kidneys and because risk of serious adverse reactions to the drug is greater in patients with impaired renal function, ZUVOG M should be used only in patients with normal renal function. Because aging is associated with reduced renal function, the use of ZUVOG M should be with caution as age increases. Care should be taken in the dose selection and regular renal function be monitored. The administration of ZUVOG M should be initiated at a lower dose in elderly patients and generally, should not be titrated to the maximum dose. The combination should be carefully administered under close observation of the course of disease conditions, with careful attention to the blood sugar level and the onset of gastrointestinal symptoms.
Renal Impairment Patients
Metformin is known to be substantially excreted by the kidney and the risk of metformin accumulation and lactic acidosis increases with the degree of impairment of renal function. Thus, patients with serum creatinine levels above the upper limit of normal for their age should not receive ZUVOG M. In patients with advanced age, ZUVOG M should be carefully titrated to establish the minimum dose for adequate glycaemic effect, because aging is associated with reduced renal function. In elderly patients, particularly those ≥80 years of age, renal function should be monitored regularly and, generally, ZUVOG M should not be titrated to the maximum dose. Before initiation of ZUVOG M and at least annually thereafter, renal function should be assessed and verified as normal. In patients in whom development of renal dysfunction is anticipated, renal function should be assessed more frequently and ZUVOG M discontinued if evidence of renal impairment is present.
4.7 Effects on ability to drive and use machines
This combination containing voglibose and metformin does not cause hypoglycaemia and therefore has no effect on the ability to drive or to use machines. However, patients should be alerted to the risk of hypoglycaemia when ZUVOG M is used in combination with other antidiabetic agents (e.g. sulphonylureas, insulin, or meglinitides).
4.8 Undesirable effects
Voglibose
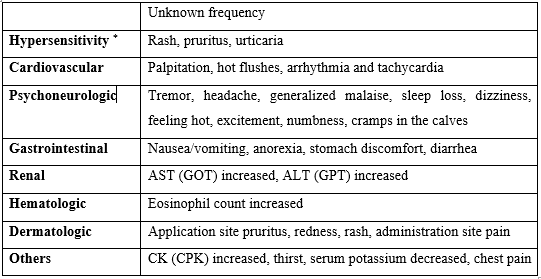
The gastrointestinal adverse effects like diarrhoea, loose stools, abdominal pain, constipation, anorexia, nausea, vomiting or heartburn may occur with the use of voglibose. Abdominal swelling, increased flatus, and intestinal obstruction like symptoms due to an increase in intestinal gas, etc. may occur. Serious hepatic dysfunction accompanied with jaundice, increased aspartate aminotransferase (AST) or alanine aminotransferase (ALT), etc. may also occur. One case of hepatitis with severe cholestasis attributed to voglibose hypersensitivity has been reported; a causal relationship appears likely. When voglibose is administered to the patients with serious liver cirrhosis, hyperammonia may worsen with the development of constipation, etc., followed by disturbance of consciousness. When voglibose is used in combination with other antidiabetic drugs, hypoglycaemia may occur.
Metformin
The most common adverse reactions of metformin hydrochloride are diarrhoea, nausea/vomiting, flatulence, asthenia, indigestion, abdominal discomfort, headache, abnormal stools, hypoglycaemia, myalgia, lightheaded, dyspnea, nail disorder, rash, sweating increased, taste disorder, chest discomfort, chills, flu syndrome, flushing and palpitation. The very rare adverse effects include isolated reports of liver function tests, abnormalities or hepatitis resolving upon metformin discontinuation and skin reactions such as erythema, pruritus, urticaria.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: https://www.zuventus.co.in/drug-safety-reporting
4.9 Overdose
Overdose of metformin hydrochloride has occurred, including ingestion of amounts greater than 50 grams. Hypoglycaemia was reported in approximately 10% of cases, but no causal association with metformin hydrochloride has been established. Lactic acidosis has been reported in approximately 32% of metformin overdose cases. Metformin is dialyzable with a clearance of up to 170 mL/min under good hemodynamic conditions. Therefore, haemodialysis may be useful for removal of accumulated drug from patients in whom metformin overdosage is suspected.
Voglibose competitively and reversibly inhibits the alpha-glucosidase enzymes in the brush border in the small intestine, which delays the hydrolysis of complex carbohydrates. It appears unlikely to produce hypoglycaemia in overdose, but abdominal discomfort and diarrhea may occur.
5.0 Pharmacological properties
5.1 Pharmacodynamic/ Mechanism of Action
Voglibose
Voglibose is an alpha glucosidase inhibitor which inhibits the activity of alpha glucosidases that catalyse the decomposition of disaccharides into monosaccharides in the intestine, thereby delaying the digestion and absorption of carbohydrates, resulting in improvement of postprandial hyperglycaemia.
Metformin Hydrochloride
Metformin improves glucose tolerance in patients with type 2 diabetes (NIDDM), lowering both basal and postprandial plasma glucose. Metformin is not chemically or pharmacologically related to sulphonylureas, thiazolidinediones, or alpha-glucosidase inhibitors. Metformin decreases hepatic glucose production, decreases intestinal absorption of glucose, and improves insulin sensitivity by increasing peripheral glucose uptake and utilization. Unlike sulphonylureas, metformin does not produce hypoglycaemia in either patient with type 2 diabetes or normal subjects and does not cause hyperinsulinaemia. With metformin therapy, insulin secretion remains unchanged while fasting insulin levels and day-long plasma insulin response may actually decrease.
5.2 Pharmacokinetic properties
Voglibose
Voglibose is poorly absorbed after oral doses. Plasma concentrations after oral doses have usually been undetectable. Following repeated administration to healthy subjects (n=6) in a single dose of 0.2 mg, 3 times a day, for 7 consecutive days, voglibose was not detected in plasma or urine. Similarly, when voglibose was administered to healthy male adults (n=10) as a single dose of 2 mg, voglibose was not detected in plasma or urine. After ingestion of voglibose, the majority of active unchanged drug remains in the lumen of the gastrointestinal tract to exert its pharmacological activity. Voglibose is metabolized by intestinal enzymes and by the microbial flora. Voglibose is mainly excreted in the feces.
Metformin Hydrochloride
The absolute bioavailability of a metformin 500-mg tablet given under fasting conditions is approximately 50-60%. Studies using single oral doses of metformin 500 mg to 1500 mg, and 850 mg to 2550 mg, indicate that there is a lack of dose proportionality with increasing doses, which is due to decreased absorption rather than an alteration in elimination. Food decreases the extent of and slightly delays the absorption of metformin, as shown by approximately a 40% lower mean peak plasma concentration (Cmax), a 25% lower area under the plasma concentration versus time curve (AUC), and a 35-minute prolongation of time to peak plasma concentration (Tmax) following administration of a single 850-mg tablet of metformin with food, compared to the same tablet strength administered fasting. The clinical relevance of these decreases is unknown.
The apparent volume of distribution (V/F) of metformin following single oral doses of metformin immediate-release 850 mg averaged 654 ± 358 L. Metformin is negligibly bound to plasma proteins, in contrast to sulphonylureas, which are more than 90% protein bound. Metformin partitions into erythrocytes, most likely as a function of time. At usual clinical doses and dosing schedules of metforminimmediate-release, steady state plasma concentrations of metformin are reached within 24-48 hours and are generally <1 µg/mL. During controlled clinical trials of immediate-release metformin, maximum metformin plasma levels did not exceed 5 µg/mL, even at maximum doses.
Intravenous single-dose studies in normal subjects demonstrate that metformin is excreted unchanged in the urine and does not undergo hepatic metabolism (no metabolites have been identified in humans) nor biliary excretion. Renal clearance of metformin is approximately 3.5 times greater than creatinine clearance, which indicates that tubular secretion is the major route of metformin elimination. Following oral administration, approximately 90% of the absorbed drug is eliminated via the renal route within the first 24 hours, with a plasma elimination half-life of approximately 6.2 hours. In blood, the elimination half-life is approximately 17.6 hours, suggesting that the erythrocyte mass may be a compartment of distribution.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
Voglibose
A study demonstrated that voglibose can potentiate CCl4 (carbon tetrachloride) and APAP (acetaminophen) hepatotoxicity in rats by inducing hepatic CYP2E1.
Metformin
Long-term carcinogenicity studies have been performed in rats (dosing duration of 104 weeks) and mice (dosing duration of 91 weeks) at doses up to and including 900 mg/kg/day and 1500 mg/kg/day, respectively. These doses are both approximately 3 times the maximum recommended human daily dose of 2550 mg based on body surface area comparisons. No evidence of carcinogenicity with metformin was found in either male or female mice. Similarly, there was no tumorigenic potential observed with metformin in male rats. There was, however, an increased incidence of benign stromal uterine polyps in female rats treated with 900 mg/kg/day.
There was no evidence of a mutagenic potential of metformin in the following in vitro tests: Ames test (S. typhimurium), gene mutation test (mouse lymphoma cells), or chromosomal aberrations test (human lymphocytes). Results in the in vivo mouse micronucleus test were also negative.
Fertility of male or female rats was unaffected by metformin when administered at doses as high as 600 mg/kg/day, which is approximately 2 times the maximum recommended human daily dose of 2550 mg based on body surface area comparisons.
7.0 Description
Zuvog M contains two oral antihyperglycemic drugs, voglibose and metformin hydrochloride, used in the management of type 2 diabetes mellitus.

8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable.
8.2 Shelf-life
Refer on pack
8.3 Packaging information
Zuvog M 0.2 / 0.3 - A blister strip of 10 tablets
8.4 Storage and handing instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children.
9.0 Patient Counselling Information
- Tablet should be swallowed whole & not to be broken, crushed or chewed.
- Take this medicine exactly as prescribed by your doctor. Do not change the dose or stop therapy without consulting your doctor.
- Tell your doctor immediately if you experience any deep or rapid breathing, persistent nausea, vomiting, and stomach pain as ZUVOG M Tablet may cause a rare but serious condition called lactic acidosis, which is an excess of lactic acid in the blood.
- Pregnant women and breastfeeding mothers should not use this medicine without doctor consultation.
- Don’t medicine for the treatment of diabetic ketoacidosis or diabetic pre-coma.
- This medicine is not advisable for use in children.
- Avoid use of this medicine during severe infection or before and after surgery or in serious trauma cases.
- This tablet may cause hypoglycemia when co-administered with oral sulfonylureas and insulin.
12.0 Date of revision
11/10/2024
The name of your medicine is ZUVOG M. We refer to them as ZUVOG M TABLETS or ZUVOG M throughout this leaflet.
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any more questions, please ask your doctor or your pharmacist.
- This medicine has been prescribed for you personally and you should not pass it on to anyone else. It may harm them, even if their symptoms are the same as yours.
- If any of the side effects get serious, or if you notice any side effects that are not listed in the leaflet, please tell your doctor or pharmacist.
In this leaflet:
1. What ZUVOG M Tablets are and what they are used for
2. What you need to know before you take ZUVOG M Tablets
3. How to take ZUVOG M Tablets
4. Possible side effects
5. How to store ZUVOG M Tablets
6. Contents of the pack and other information
1. What ZUVOG M Tablets are and What they are used for
ZUVOG M contains two active substances, voglibose and metformin hydrochloride. It is used as a second-line treatment for type II diabetes when diet, exercise, and either voglibose or metformin alone have not been enough to control blood glucose (sugar).
Voglibose works by blocking certain enzymes in your small intestine that break down complex sugars into simple sugars like glucose. By doing this, it slows down the absorption of sugar into your bloodstream after meals, helping to control blood sugar levels.
Insulin is a hormone that enables body tissues to take glucose from the blood and to use it for energy or for storage for future use. People with Type 2 diabetes do not make enough insulin in their pancreas or their body does not respond properly to the insulin it does make. This causes a build-up of glucose in the blood which can cause a number of serious long-term problems so it is important that you continue to take your medicine, even though you may not have any obvious symptoms. Metformin hydrochloride makes the body more sensitive to insulin and helps return to normal the way your body uses glucose.
2. What You Need to Know Before You Take Zuvog M Tablets
Do not take ZUVOG M if you:
- Are allergic to Metformin, Voglibose or any of the other ingredients of this medicine.
- Have inflammatory bowel disease, gastrointestinal obstruction, or conditions that may deteriorate due to increased gas formation.
- Have severe ketosis, diabetic coma or pre-coma, severe infection, or hepatic impairment.
- Have kidney disease or dysfunction.
- Are undergoing radiologic studies involving iodinated contrast materials.
- you have been treated for acute heart problems or have recently had a heart attack or have severe circulatory problems or breathing difficulties. This may lead to a lack in oxygen supply to tissue which can put you at risk for lactic acidosis
- you are a heavy drinker of alcohol.
- you are under 18 years of age.
Warnings and precautions
Risk of lactic acidosis
ZUVOG M Tablets may cause a very rare, but very serious side effect called lactic acidosis, particularly if your kidneys are not working properly. The risk of developing lactic acidosis is also increased with uncontrolled diabetes, serious infections, prolonged fasting or alcohol intake, dehydration, liver problems and any medical conditions in which a part of the body has a reduced supply of oxygen (such as acute severe heart disease).
If any of the above apply to you, talk to your doctor for further instructions.
Stop taking ZUVOG M Tablets and contact a doctor or the nearest hospital immediately if you experience some of the symptoms of lactic acidosis, as this condition may lead to coma.
Symptoms of lactic acidosis include:
- Vomiting
- stomach-ache (abdominal pain)
- muscle cramps
- a general feeling of not being well with severe tiredness
- difficulty in breathing
- reduced body temperature and heartbeat
Lactic acidosis is a medical emergency and must be treated in a hospital
Taking other medicines and ZUVOG M
- Antidiabetic Medications: Co-administration with sulfonylureas or insulin can enhance the hypoglycemic effect, increasing the risk of hypoglycemia. Close monitoring of blood glucose levels is recommended.
- Beta Blockers: These medications can mask the symptoms of hypoglycemia, making it harder to recognize low blood sugar levels.
- Keratolytics and MAOIs: These can enhance the hypoglycemic effect of Voglibose.
- Anti-lipidemic Agents: These can enhance the hypoglycemic effect of Voglibose.
- Sympathomimetics or Adrenergic Agonists: These can reduce the hypoglycemic effect of Voglibose.
- Adrenocorticotropic and Thyroid Hormones: These can reduce the hypoglycemic effect of Voglibose.
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
It is especially important to mention the following:
- Medicines which increase urine production (diuretics (water tablets) such as furosemide)
- medicines used to treat pain and inflammation (NSAID and COX-2-inhibitors, such as ibuprofen and celecoxib)
- certain medicines for the treatment of high blood pressure (ACE inhibitors and angiotensin II receptor antagonists)
- Steroids such as prednisolone, mometasone, beclometasone.
- Sympathomimetic medicines including epinephrine and dopamine used to treat heart attacks and low blood pressure. Epinephrine is also included in some dental anaesthetics.
- Medicines that may change the amount of metformin in your blood, especially if you have reduced kidney function (such as verapamil, rifampicin, cimetidine, dolutegravir, ranolazine, trimethoprim, vandetanib, isavuconazole, crizotinib, olaparib).
ZUVOG M Tablets with alcohol
Avoid excessive alcohol intake while taking Metformin Hydrochloride Tablets since this may increase the risk of lactic acidosis.
Pregnancy
ZUVOG M is generally not recommended during pregnancy due to insufficient well-controlled studies in pregnant women. It should only be used if specifically advised by a healthcare professional in rare, carefully evaluated situations.
Breast-feeding
The use of ZUVOG M during breastfeeding is not well-studied, and there is limited data available on its safety for nursing mothers and infants. It is generally recommended to avoid ZUVOG M during breastfeeding unless absolutely necessary.
Driving and using machines
ZUVOG M does not usually affect your ability to drive or use machines. However, if you experience symptoms of hypoglycemia, such as dizziness or confusion, avoid these activities until you feel better.
3. How to Take Zuvog M Tablets
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
Dosage:
The initial recommended dose: one tablet of ZUVOG M 0.2 three times daily just before each meal.
If the effect is inadequate, the dose may be increased to one tablet of ZUVOG M 0.3 three times daily just before each meal.
Method of administration:
ZUVOG-M Tablet should be taken with food. Take it regularly at the same time each day to get the most benefit.
If you take more ZUVOG M Tablets than you should
Taking more Zuvog M Tablets than prescribed can lead to an overdose. While ZUVOG M is unlikely to cause hypoglycemia (low blood sugar) in overdose, it can cause significant gastrointestinal discomfort. Abdominal discomfort, diarrhea, Flatulence and Bloating may occur. Do not panic. Stay calm and seek medical advice immediately. Contact your doctor or go to the nearest hospital emergency department. Take the medicine packaging with you to show the healthcare professionals. Treatment is generally supportive and symptomatic, focusing on relieving gastrointestinal symptoms.
If you forget to take Zuvog M Tablets
If you forget to take a tablet, take one as soon as you remember, unless it is nearly time to take the next one. Never take two doses together. Take the remaining doses at the correct time.
If you stop taking Zuvog M Tablets
Take this medicine for as long as your doctor tells you to, as you may become unwell if you stop.
4. Possible Side Effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Voglibose
- Gastrointestinal Issues: Diarrhea, loose stools, abdominal pain, constipation, anorexia, nausea, vomiting, and heartburn.
- Abdominal Swelling and Increased Flatus: Symptoms similar to intestinal obstruction due to increased intestinal gas.
- Serious Hepatic Dysfunction: Jaundice, increased levels of liver enzymes (AST or ALT). One case of hepatitis with severe cholestasis attributed to voglibose hypersensitivity has been reported.
- Hyperammonemia: In patients with serious liver cirrhosis, voglibose may worsen hyperammonemia, leading to constipation and disturbance of consciousness.
- Hypoglycemia: When used in combination with other antidiabetic drugs.
Metformin:
- Common Adverse Reactions: Diarrhea, nausea/vomiting, flatulence, asthenia (weakness), indigestion, abdominal discomfort, headache, abnormal stools, hypoglycemia, myalgia (muscle pain), lightheadedness, dyspnea (difficulty breathing), nail disorder, rash, increased sweating, taste disorder, chest discomfort, chills, flu syndrome, flushing, and palpitations.
- Very Rare Adverse Effects: Liver function test abnormalities or hepatitis resolving upon discontinuation of metformin, and skin reactions such as erythema (redness of the skin), pruritus (itching), and urticaria (hives).
- Serious side effects: Lactic acidosis, characterized by symptoms such as deep or rapid breathing, persistent nausea, vomiting, and stomach pain.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly:
Website: www.zuventus.com and click the tab “Safety Reporting” located on the top t end of the home page.
Website link: https://www.zuventus.co.in/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
You can also report the side effect with the help of your treating physician.
5. How to Store Zuvog M Tablets
Do not use the tablets after the end of the expiry month (use-by date) shown on the product packaging.
Store below 30°C. Protected from moisture.
Keep this medicine out of the sight and reach of children
6. Contents of the Pack and Other Information
What ZUVOG M Tablets contain
The active substances are Voglibose and Metformin Hydrochloride.
Each uncoated bilayered tablet contains:
Voglibose IP………………………0.2 mg/ 0.3 mg
Metformin Hydrochloride IP……...500 mg
(As Prolonged-release form)
Excipients……………………………. q.s.
Packaging:
ZUVOG M 0.2/0.3: A Blister strips of 10 tablets