Respilax Capsules


1.0 Generic name
Doxycycline & Lactic Acid Bacillus Capsules
2.0 Qualitative and quantitative composition
Each hard gelatin capsule contains :
Doxycycline Hyclate IP
equivalent to Doxycycline 100 mg
(as immediate-release pellets)
Lactic acid bacillus 5 billion spores
(as enteric coated pellets)
Colour : Titanium Dioxide IP
Excipients q.s.
Approved colours used in the capsule shell.
3.0 Dosage form and strength
Capsule
4.0 Clinical particulars
4.1 Therapeutic indication
- For adult patients prone to intrabdominal bacterial infection & Antibiotic associated diarrhoea.
- Respiratory tract infections
- Urinary tract infections
- Sexually transmitted diseases
- Dermatological infections
- Ophthalmic infections
- Rickettsial infections
Other infections
Doxycycline Capsules are indicated for prophylaxis in the
following conditions : Scrub typhus, Travellers' diarrhoea, Leptospirosis.
4.2 Posology and method of administration
The capsules should be swallowed with plenty of fluid in either the resting or standing position and well before going to bed for the night to reduce the likelihood of oesophageal irritation and ulceration. If gastric irritation occurs, it is recommended that Doxycycline Capsules be given with food or milk. Studies indicate that the absorption of Doxycycline is not notably influenced by simultaneous ingestion of food or milk.
Posology
The usual dosage of Doxycycline for the treatment of acute infections in adults is 200 mg on the first day (as a single dose or in divided doses with a twelve-hour interval) followed by a maintenance dose of 100mg/day. In the management of more severe infections (particularly chronic infections of the urinary tract), 200 mg daily should be given throughout the treatment period. Exceeding the recommended dosage may result in an increased incidence of side effects. Therapy should be continued for at least 24 to 48 hours after the symptoms and fever have subsided. When used in streptococcal infections, therapy should be continued for 10 days to prevent the development of rheumatic fever or glomerulonephritis.
Dosage recommendations in specific infections :
Acne vulgaris
50 mg daily with food or fluid for 6 to 12 weeks.
Sexually transmitted diseases
100 mg twice daily for 7 days is recommended in the following infections : uncomplicated gonococcal infections (except anorectal infections in men); uncomplicated urethral, endocervical or rectal infection caused by Chlamydia trachomatis; non-gonococcal urethritis caused by Ureaplasma urealyticum. Acute epididymo-orchitis caused by Chlamydia trachomatis or Neisseria gonorrhoea 100 mg twice daily for 10 days.
Primary and secondary syphilis
300 mg a day in divided doses for at least 10 days.
Louse and tick-borne relapsing fevers
A single dose of 100mg or 200mg according to severity.
Treatment of chloroquine-resistant falciparum malaria
200 mg daily for at least 7 days. Due to the potential severity of the infection, a rapid- acting schizonticide such as quinine should always be given in conjunction with Doxycycline; quinine dosage recommendations vary in different areas
Prophylaxis of malaria
100 mg daily in adults and children over the age of 12 years. Prophylaxis can begin 1 - 2 days before travel to malarial areas. It should be continued daily during travel in the malarial areas and for 4 weeks after the traveller leaves the malarial area. For current advice on geographical resistance patterns and appropriate chemoprophylaxis, current guidelines or the Malaria Reference Laboratory should be consulted, details of which can be found in the British National Formulary (BNF).
For the prevention of scrub typhus
200 mg as a single dose.
For the prevention of travellers' diarrhoea in adults
200 mg on the first day of travel (administered as a single dose or as 100 mg every 12 hours) followed by 100 mg daily throughout the stay in the area. Data on the use of the drug prophylactically are not available beyond 21 days.
For the prevention of leptospirosis
200 mg once each week throughout the stay in the area and 200 mg at the completion of the trip. Data on the use of the drug prophylactically are not available beyond 21 days
Children aged 8 years to less than 12 years
The use of Doxycycline for the treatment of acute infections in children aged 8 years to less than 12 years should be carefully justified in situations where other drugs are not available, are not likely to be effective or are contraindicated.
In such circumstance, the doses for the treatment of acute infections are :
For children 45 kg or less
Initial dose : 4.4 mg/kg (in single or 2 divided doses) with maintenance dose : 2.2 mg/kg (in single or 2 divided doses). In the management of more severe infections, up to 4.4 mg/kg should be given throughout treatment.
For children, over 45 kg
Dose administered for adults should be used.
Children aged from birth to less than 8 years
Doxycycline should not be used in children aged younger than 8 years due to the risk of teeth discolouration.
Paediatric population
Not recommended
Elderly patients
Doxycycline may be prescribed in the usual dose with no special precautions. No dosage adjustment is necessary in the presence of renal impairment.
Renal impairment : Studies to date have indicated that administration of Doxycycline at the usual recommended doses does not lead to excessive accumulation of the antibiotic in patients with renal impairment.
The anti-anabolic action of the Tetracycline may cause an increase in blood urea. Studies to date indicate that this does not occur with the use of Doxycycline in patients with impaired renal function. Haemodialysis does not alter the serum half-life of Doxycycline.
4.3 Contraindications
• Hypersensitivity to the active substance, any of the tetracycline or to any of the excipients listed in the formulation.
• Sucrose intolerance : Patients with rare hereditary problems of fructose intolerance, glucose galactose malabsorption or sucrose-isomaltase insufficiency should not take Doxycycline.
4.4 Special warnings and precautions for use
Paediatric population
The use of drugs of the tetracycline class during tooth development (last half of pregnancy; infancy and childhood to the age of 8 years) may cause permanent discolouration of the teeth (yellow-grey-brown). This adverse reaction is more common during long-term use of the drugs but has been observed following repeated short-term courses. Enamel hypoplasia has also been reported. Use Doxycycline in paediatric patients aged younger than 8 years only when the potential benefits are expected to outweigh the risks in severe or life- threatening conditions (e.g. Rocky Mountain spotted fever), particularly only when there are no adequate alternative therapies. Although the risk of permanent teeth staining is rare in children aged 8 years to less than 12 years, the use of Doxycycline should be carefully justified in situations where other drugs are not available, are not likely to be effective or are contraindicated.
Photosensitivity
Photosensitivity manifested by an exaggerated sunburn reaction has been observed in some individuals taking Tetracycline, including Doxycycline. Patients likely to be exposed to direct sunlight or ultraviolet light should be advised that this reaction can occur with tetracycline drugs and treatment should be discontinued at the first evidence of skin erythema. Photoonycholysis has also been reported in patients receiving Doxycycline.
Use in patients with impaired hepatic function
Doxycycline should be administered with caution to patients with hepatic impairment or those receiving potentially hepatotoxic drugs. Abnormal hepatic function has been reported rarely and has been caused by both the oral and parenteral administration of Tetracycline, including Doxycycline.
Use in patients with renal impairment
Excretion of Doxycycline by the kidney is about 40%/72 hours in individuals with normal renal function. This percentage excretion may fall to a range as low as 1-5%/72 hours in individuals with severe renal insufficiency (creatinine clearance below 10ml/min). Studies have shown no significant difference in the serum half-life of Doxycycline in individuals with normal and severely impaired renal function. Haemodialysis does not alter the serum half-life of Doxycycline. The anti-anabolic action of the Tetracyclines may cause an increase in blood urea. Studies to date indicate that this anti-anabolic effect does not occur with the use of Doxycycline in patients with impaired renal function.
Serious skin reactions
Serious skin reactions, such as exfoliative dermatitis, erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported in patients receiving Doxycycline. If serious skin reactions occur, Doxycycline should be discontinued immediately and appropriate therapy should be instituted.
Microbiological overgrowth
The use of antibiotics may occasionally result in overgrowth of non-susceptible organisms including Candida. If a resistant organism appears, the antibiotic should be discontinued and appropriate therapy instituted.
Pseudomembranous colitis has been reported with nearly all antibacterial agents, including Doxycycline, and has ranged in severity from mild to life-threatening. It is important to consider this diagnosis in patients who present with diarrhoea subsequent to the administration of antibacterial agents. Clostridium difficile associated diarrhoea (CDAD) has been reported with use of nearly all antibiotics, including Doxycycline, and has ranged in severity from mild diarrhoea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C difficile. C difficile produces toxins A and B, which contribute to development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD should be considered in all patients who present with diarrhoea after antibiotic treatment. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
Oesophagitis
Instances of oesophagitis and oesophageal ulcerations have been reported in patients receiving capsule and tablet forms of drugs in the tetracycline class, including Doxycycline. Most of these patients took medications immediately before going to bed or with inadequate amounts of fluid.
Benign intracranial hypertension
Bulging fontanelles in infants have been reported in individuals receiving Tetracyclines. Benign intracranial hypertension (pseudotumor cerebri) has been associated with the use of Tetracyclines including Doxycycline. Benign intracranial hypertension (pseudotumor cerebri) is usually transient, however cases of permanent visual loss secondary to benign intracranial hypertension (pseudotumor cerebri) have been reported with Tetracyclines including Doxycycline. If visual disturbance occurs during treatment, prompt ophthalmologic evaluation is warranted. Since intracranial pressure can remain elevated for weeks after drug cessation patients should be monitored until they stabilize. Concomitant use of isotretinoin or other systemic retinoids and Doxycycline should be avoided because isotretinoin is also known to cause benign intracranial hypertension (pseudotumor cerebri).
Porphyria
There have been rare reports of porphyria in patients receiving Tetracyclines
Venereal disease
When treating venereal disease, where co-existent syphilis is suspected, proper diagnostic procedures, including dark-field examinations, should be utilised. In such cases monthly serological tests should be performed for at least four months.
Beta-haemolytic streptococci infections
Infections due to Group A beta-haemolytic streptococci should be treated for at least 10 days
Myasthenia gravis
Due to a potential for weak neuromuscular blockade, care should be taken in administering Tetracyclines to patients with myasthenia gravis.
Systemic lupus erythematosus
Tetracyclines can cause exacerbation of systemic lupus erythematosus (SLE).
Jarisch-Herxheimer reaction
Some patients with spirochete infections may experience a Jarisch-Herxheimer reaction shortly after Doxycycline treatment is started. Patients should be reassured that this is a usually self-limiting consequence of antibiotic treatment of spirochete infections
Methoxyflurane
Caution is advised in administering Tetracyclines with Methoxyflurane
4.5 Drugs interactions
The absorption of Doxycycline may be impaired by concurrently administered antacids containing aluminium, calcium, magnesium or other drugs containing these cations; Oral Zinc, Iron salts or Bismuth preparations. Dosages should be maximally separated. Since bacteriostatic drugs may interfere with the bactericidal action of penicillin, it is advisable to avoid giving Doxycycline in conjunction with penicillin. There have been reports of prolonged prothrombin time in patients taking warfarin and Doxycycline. Tetracyclines depress plasma prothrombin activity and reduced doses of concomitant anticoagulants may be necessary.
The serum half-life of Doxycycline may be shortened when patients are concurrently receiving Barbiturates, Carbamazepine or Phenytoin. An increase in the daily dosage of Doxycycline should be considered
Alcohol may decrease the half-life of Doxycycline
A few cases of pregnancy or breakthrough bleeding have been attributed to the concurrent use of tetracycline antibiotics with oral contraceptives. Doxycycline may increase the plasma concentration of Ciclosporin. Co-administration should only be undertaken with appropriate monitoring. The concurrent use of Tetracyclines and Methoxyflurane has been reported to result in fatal renal toxicity. Concomitant use of isotretinoin or other systemic retinoids and Doxycycline should be avoided. Each of these agents used alone has been associated with benign intracranial hypertension (pseudotumor cerebri).
Laboratory test interactions
False elevations of urinary catecholamine levels may occur due to interference with the fluorescence test. Drugs that induce hepatic enzymes such as Rifampicin may accelerate the decomposition of Doxycycline, thereby decreasing its half-life. Sub-therapeutic Doxycycline concentrations may result. Monitoring concurrent use is advised and an increase in Doxycycline dose may be required.
Ergotamine : There is an increased risk of Ergotism when Doxycycline is co-administered with Ergotamine.
Methotrexate : Doxycycline increases the risk of Methotrexate toxicity; prescribe with caution to patients on Methotrexate.
Quinapril contains Magnesium Carbonate and may interfere with the absorption of Doxycycline
4.6 Use in special populations
Pregnancy
Doxycycline is contraindicated in pregnancy. It appears that the risks associated with the use of Tetracyclines during pregnancy are predominantly due to effects on teeth and skeletal development.
Nursing mothers
Tetracyclines are excreted into milk and are therefore contraindicated in nursing mothers.
4.7 Effects on ability to drive and use machines
Visual disturbances such as blurring of vision may occur during treatment with Doxycycline and in such cases; patients must refrain from driving or operating machinery.
4.8 Undesirable effect
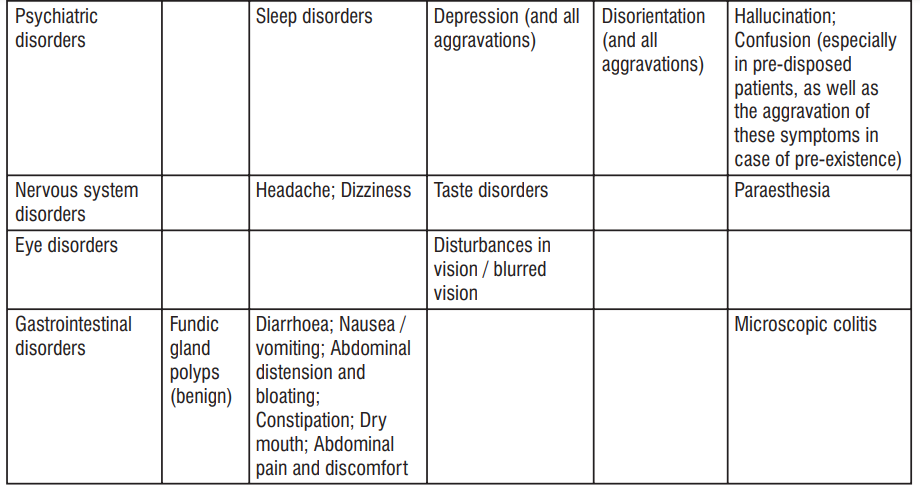
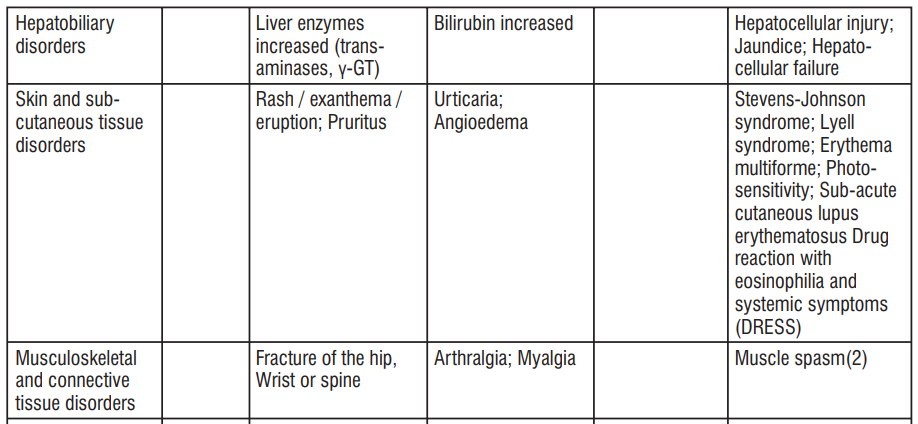
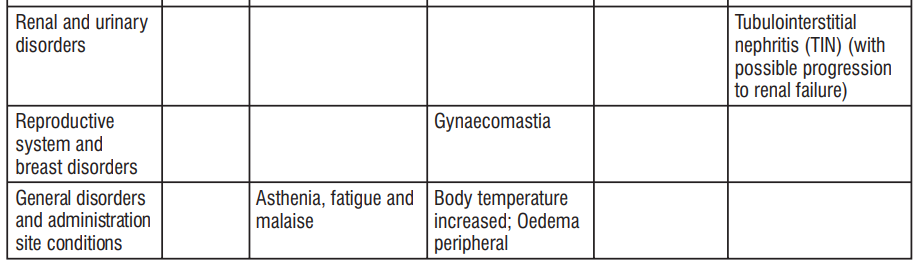
| System Organ Class | Common ≥ 1/100 to < 1/10 | Uncommon ≥ 1/1000 to < 1/100 | Rare ≥ 1/10000 to <1/1000 | Not known Cannot be estimated from the available data |
| Infections and infestations | Vaginal infection | Candida Infection, Pseudomembranous colitis, Clostridium difcile colitis | ||
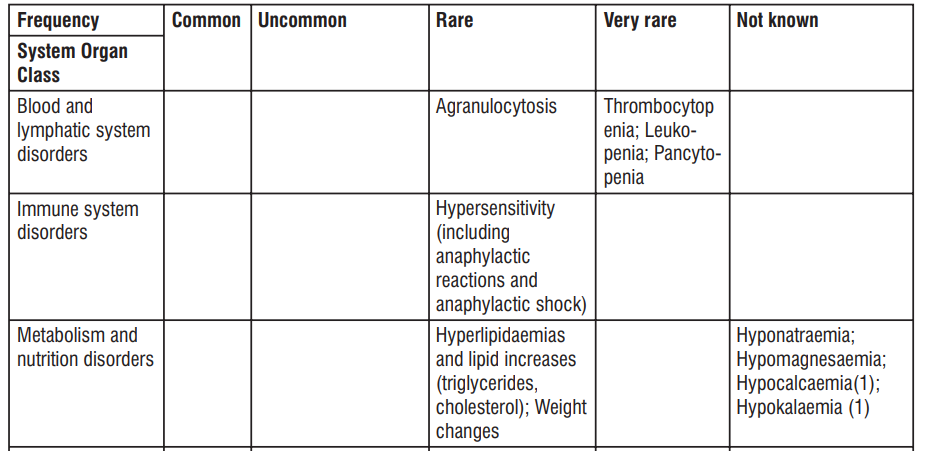
| Blood and lymphatic system disorders | Haemolyticanaemia, Neutropenia, Thrombocytopenia, Eosinophilia | |||
| Immune system disorders | Hypersensitivity (including anaphylactic shock, Anaphylactic reaction, anaphylactoid reaction, Exacerbation of systemic lupus erythematosus, Serum sickness) | Jarisch- Herxheimer reaction | ||
| Congenital, familial and genetic disorders | Porphyria | |||
| Endocrine disorders | Brown-black microscopic discolouration of thyroid glands | |||
| Metabolism and nutrition disorders | Decreased appetite | |||
| Nervous system disorders | Headache | Benign intracranial hypertension (pseudotumor cerebri), Fontanelle bulging | ||
| Psychiatric Disorders | Anxiety | |||
| Ear and labyrinth Disorders | Tinnitus | |||
| Vascular disorders | Hypotension | Flushing | ||
| Gastrointestinal disorders | Nausea/vomiting | Dyspepsia (Heartburn/ gastritis) | Pancreatitis, oesophageal ulcer, Oesophagitis, Enterocolitis, inammatory lesions (with monilial overgrowth) in the anogenital region, Dysphagia, abdominal pain, Diarrhoea, Glossitis, Stomatitis | Tooth discolouration |
| Hepatobiliary disorders | Photosensitivity reaction, rash including maculopapular and erythematous rashes, Henoch Schonlein purpura, Urticaria. | Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), Angioedema, Toxic epidermal necrolysis, Stevens Johnson syndrome, Erythema multiforme, Dermatitis exfoliative, Photoonycholysis, Skin hyperpigmentation, Fixed drug eruption (FDE) | ||
| Musculoskeletal, connective tissue and bone disorders | Arthralgia, Myalgia | |||
| Renal and urinary disorders | Blood urea increased | |||
| Cardiac Disorders | Pericarditis, Tachycardia | |||
| Respiratory, thoracic and mediastinal disorders | Dyspnoea | |||
| General disorders and administration site conditions | Peripheral oedema |
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : http://www.zuventus.co.in/safety.aspx By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Acute overdosage with antibiotics is rare. In the event of overdosage gastric lavage plus appropriate supportive treatment is indicated. Dialysis does not alter serum half-life and thus would not be of benefit in treating cases of overdosage.
5.0 Pharmacological properties
5.1 Mechanism of action
The main mechanism of action of Doxycycline is on protein synthesis. Doxycycline passes directly through the lipid bilayer of the bacterial cell wall and an energy dependent active transport system pumps the drug through the inner cytoplasmic membrane. Once inside the cell Doxycycline inhibits protein synthesis by binding to 30S ribosomes and prevents the addition of amino acids to the growing peptide chain. Doxycycline will impair protein synthesis in mammalian cells at very high concentrations but these cells lack the active transport system found in bacteria. Doxycycline is clinically effective in the treatment of a variety of infections caused by a wide range of gramnegative and gram-positive bacteria, as well as certain other micro- organisms
Lactic Acid bacillus spores, helps re-establish normal condition in the colon and has been shown to protect against antibiotic induced diarrhoea.
5.2 Pharmacodynamic properties
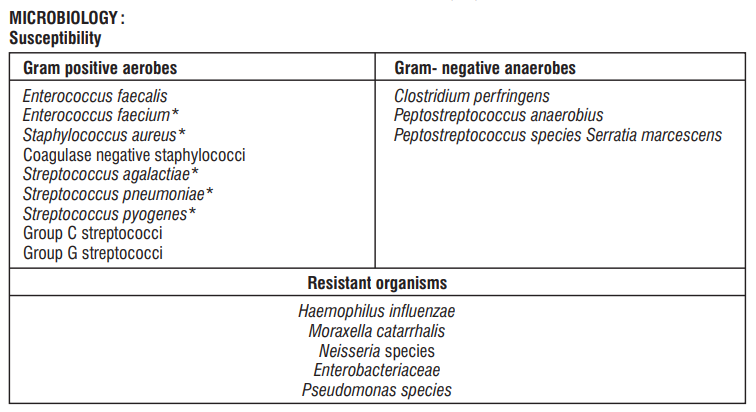
Doxycycline is clinically effective in the treatment of a variety of infections caused by a wide range of gramnegative and gram-positive bacteria, as well as certain other micro- organisms. Antimicrobial Spectrum
Gram-negative Bacteria
Acinetobacter species; Bartonella bacilliformis; Brucella species; Campylobacter fetus; Enterobacter aerogenes; Escherichia coli; Francisella tularensis; Haemophilus ducreyi; Haemophilus inuenza; Klebsiella granulomatis; Klebsiella species; Neisseria gonorrhoeae; Shigella species; Vibrio cholerae; Yersinia pestis.
Gram-positive Bacteria
Bacillus anthracis; Streptococcus pneumoniae.
Anerobic Bacteria
Clostridium species; Fusobacterium fusiforme; Propionibacterium acnes
Other Bacteria
Norcardiae and other aerobic Actinomyces species; Borrelia recurrentis; Chlamydophila psittaci; Chlamydia trachomatis; Mycoplasma pneumonia; Rickettsiae; Treponema pallidum; Treponema pallidum subspecies pertenue; Ureaplasma urealyticum.
Parasites
Balantidium coli; Entamoeba species; Plasmodium falciparum. Lactic acid bacillus is a pro biotic. It is known to have the following actions. Prevention and reduction of intestinal tract infections Enhances immune response Maintains mucosal integrity
5.3 Pharmacokinetic properties
Absorption
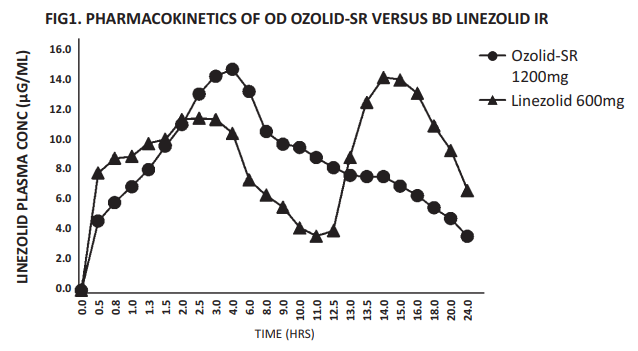
Doxycycline is almost completely absorbed and is not subject to presystemic metabolism, the mean bioavailability being approximately 93%. Absorption is rapid (effective concentrations are attained as from the first hour), and the peak serum concentration occurs after 2 to 4 hours. Almost all of the product is absorbed in the upper part of the digestive tract. Absorption is not modied by administration with meals, and milk has little effect.
Distribution
Tissue distribution is good and Doxycycline has a strong affinity for renal and lung tissue. The volume of distribution for Doxycycline ranges from 0.9 - 1.8 lkg-1.
In adults, an oral dose of 200 mg results in;
• A peak serum concentration of more than 3 μg/ml.
• A residual concentration of more than 1 μg/ml after 24 hours.
• A serum half-life of 16 to 22 hours.
Protein binding varying between 82 and 93% (labile binding) intra- and extracellular diffusion is good. With usual dosages, effective concentrations are found in the ovaries, uterine tubes, uterus, placenta, testicles, prostate, bladder, kidneys, lung tissue, skin, muscles, lymph glands, sinus secretions, maxillary sinus, nasal polyps, tonsils, liver, hepatic and gallbladder bile, gallbladder, stomach, appendix, intestine, omentum, saliva and gingival uid. Doxycycline is transferred into breast milk. Only small amounts are diffused into the cerebrospinal uid.
Biotransformation
No significant metabolism occurs.
Elimination
Doxycycline is cleared intact by renal and biliary mechanisms The antibiotic is concentrated in the bile. About 40% of the administered dose is eliminated in 3 days in active form in the urine and about 32% in the faeces. Urinary concentrations are roughly 10 times higher than plasma concentrations at the same time. In the presence of impaired renal function, urinary elimination decreases, faecal elimination increases and the half-life remains unchanged. The half-life is not affected by haemodialysis. Lactic acid bacillus, is a non pathogenic live spores which can germinate in intestine, survives gastric acidity and proliferate with inherent potentiality.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
No known animal toxicology data.
7.0 Description
Doxycycline is a broad-spectrum antibacterial. It is derived synthetically from Oxytetracycline. Lactic acid bacillus is a non-pathogenic, gram-positive, spore-forming, Lactic-acid producing bacillus. It forms Lactic acid as the main end product of Carbohydrate metabolism. They are commercialized as probiotics. In humans, they live as commensals in oral cavities, gastrointestinal tracts (GIT), and vagina.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf life
Refer on the pack.
8.3 Packaging information
Alu strip of 10 capsules.
8.4 Storage and handling instructions
Store below 25°C. Protect from light & moisture.
Keep out of reach of children.
Capsule should be swallowed whole & not to be opened, chewed or crushed.
9.0 Patient counselling information
All patients taking Doxycycline should be advised
- To avoid excessive sunlight or artificial ultraviolet light while receiving Doxycycline and to discontinue therapy if phototoxicity (e.g., skin eruption, etc.) occurs. Sunscreen or sunblock should be considered.
- To drink fluids liberally along with Doxycycline to reduce the risk of esophageal irritation and ulceration.
- That the absorption of Tetracyclines is reduced when taken with foods, especially those which contain calcium. However, the absorption of Doxycycline is not markedly influenced by simultaneous ingestion of food or milk.
- That the absorption of Tetracyclines is reduced when taking bismuth subsalicylate.
- That the use of Doxycycline might increase the incidence of vaginal candidiasis.
Patients should be counselled that antibacterial drugs, including Doxycycline should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When Doxycycline is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by Doxycycline or other antibacterial drugs in the future. Diarrhea is a common problem caused by antibacterial drugs, which usually ends when the antibacterials are discontinued. Sometimes after starting treatment with antibacterial drugs, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibacterial drug. If this occurs, patients should contact their physician as soon as possible.
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What Respilax is and what it is used for
- What you need to know before you take Respilax
- How to take Respilax
- Possible side effects
- How to store Respilax
- Contents of the pack and other information
1. What Respilax is and what is it used for
Respilax a combination medicine that contains the active substance doxycycline as doxycycline hyclate and a probiotic, lactic acid bacillus (5 billion spores). Respilax (Doxycycline) is an antibiotic belonging to a group of medicines called tetracyclines.
Doxycycline is an antibiotic which stops bacterial growth by preventing the synthesis of essential proteins required by the bacteria to carry out vital functions. lactic acid bacillus is a live microorganism (probiotics) which restores the balance of good bacteria in the intestine that may get upset with antibiotic use or due to intestinal infections. It also prevents diarrhea which may occur as side effect of this medicine (doxycycline).
Respilax capsule is used to treat many different types of infections including:
- Chest, lung or nasal infections e.g. bronchitis, pneumonia, sinusitis
- Urinary tract infections (the passage through which urine passes) e.g. cystitis, urethritis
- Acne (a skin condition), Dermatological infections
- Eye infections
- Sexually transmitted diseases e.g. gonorrhoea, syphilis, chlamydia
- Fevers associated with louse or tick bites
- Malaria, when chloroquine is not effective
- For the prevention of travellers' diarrhea in adults
Respilax is also used to prevent certain infections developing, these are scrub typhus (a disease carried by small insects) Rocky Mountain spotted fever, travellers’ diarrhoea, malaria and leptospirosis (a bacterial infection).
Your doctor may want you to take Respilax to treat another infection not listed above. You may also be prescribed an additional medicine to take with Respilax to treat your infection. You must talk to your doctor if you do not feel better or if you feel worse.
2. What you need to know before you take Respilax
Do not take Respilax:
- if you are allergic to doxycycline or any other tetracycline antibiotic or any of the other ingredients of this medicine (listed in section 6)
- if you are pregnant or trying to become pregnant
- if you are breast feeding
You should not use Respilax during periods of tooth development (pregnancy, infancy or in children below 8 years old) as such use may lead to permanent discoloration (yellow-grey-brown).
There may be circumstances (e.g. severe or life-threatening conditions), where your physician may decide that the benefits outweigh this risk in children below 8 years and Respilax should be prescribed.
Warnings and precautions
Talk to your doctor or pharmacist before taking Respilax if any of the following apply to you:
- you are likely to be exposed to strong sunlight or UV light (e.g. on a sun bed). You should avoid exposure to strong sunlight while taking this medicine as your skin may be more sensitive to sunburn than normal.
- you have kidney or liver problems.
- you have myasthenia gravis (a disease which causes unusual tiredness and weakness of certain muscles, particularly in the eyelid).
- you have an immune system disease that causes joint pain, skin rashes and fever (systemic lupus erythematosus).
- The condition may be worsened by taking Respilax.
- you have porphyria (a rare disease of blood pigments).
- you have (or have ever had) systemic lupus erythematosus (an allergic condition that causes joint pain, skin rashes and fever).
- This condition may be worsened by taking Respilax. you are suspected as having syphilis.
- Your doctor will continue to monitor you after your treatment has stopped.
- you have diarrhoea or usually get diarrhoea when you take antibiotics or have suffered from problems with your stomach or intestines.
- If you develop severe or prolonged or bloody diarrhoea during or after using Respilax tell your doctor immediately since it may be necessary to interrupt the treatment.
- This may be a sign of bowel inflammation (pseudomembranous colitis) which can occur following treatment with antibiotics.
- you are taking oral retinoids as there is a higher risk of suffering from increased pressure in your skull (severe headache with change in vision) when taken with Respilax.
When used for a long duration, Respilax may cause infections that cannot be treated with this antibiotic. Your doctor can explain the signs and symptoms of such types of infection.
Other medicines and Respilax
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines (including medicines you have obtained without a prescription).
If you are taking any of the medicines listed below tell your doctor before taking Respilax.
Some medicines can reduce the effectiveness of Respilax, these include:
- Antacids (indigestion remedies), iron preparations, oral zinc or bismuth. These should not be taken at the same time of day as Respilax.
- Carbamazepine, phenytoin (medicines used to control epilepsy) and barbiturates (used to control epilepsy or as a sedative).
Respilax can affect the action of some other medicines, these include:
- Increased action of warfarin or coumarins (used to prevent blood clots)
- Reduced effectiveness of oral contraceptives (birth control pills)
- Reduced effectiveness of penicillin antibiotics (used to treat infections)
- Increased blood levels of ciclosporin (a medicine used to affect the body’s immune response)
If you are going to have a general anaesthetic for an operation or dental surgery you must tell your anaesthetist or dentist that you are taking doxycycline as you may have more side effects.
Taking Respilax with food, drink and alcohol
Please see section 3 How to take Respilax
Alcohol may reduce the effect of Respilax and should be avoided.
Pregnancy and breast-feeding
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby ask your doctor or pharmacist for advice before taking this medicine. Respilax must not be taken if you are pregnant or breast-feeding.
Driving and using machines
This medicine should not affect your ability to drive or use machines.
3. How to take Respilax
Always take this medicine exactly as your doctor or pharmacist has told you to. Check with your doctor or pharmacist if you are not sure.
- The capsules should be swallowed with plenty of water.
- It is best to take your capsules at the same time(s) each day, when sitting or while standing.
- It is important not to lie down for at least thirty minutes after taking Doxycycline capsules, so that the capsule can move as swiftly as possible into the stomach and prevent irritation of the throat oro-esophagus (canal taking food from the mouth to the stomach).
- For the treatment of infections, Doxycycline capsules can be taken with or without food.
- For the treatment of acne, it is recommended to take Doxycycline capsules with food or a drink.
- If Doxycycline capsules upset your stomach, then taking it with food or milk is recommended.
Usual Dose (Chest, lung or nasal, urinary tract, eye and other infections)
The recommended doses are shown in the list below. These are the different doses that your doctor may prescribe depending on the infection being treated.
2 Capsules on the first day, then 1 capsule daily. The length of treatment is dependent on the infection being treated.
Children aged 8 years to less than 12 years
Doxycycline for the treatment of acute infections in children aged 8 years to less than 12 years should be used in situations where other drugs are not available or are not likely to be effective. In such circumstances, the usual doses are:
For children 45 kg or less
First day: 4.4 mg for each kg of bodyweight (in single or 2 divided doses) then 2.2 mg for each kg of bodyweight (in single or 2 divided doses) from the second day. The length of treatment is dependent on the infection being treated.
In more severe infections, up to 4.4 mg for each kg of bodyweight should be given throughout treatment.
For children over 45 kg
Dose administered for adults should be used; 200mg on the first day, then 100 mg daily. The length of treatment is dependent on the infection being treated.
Adults and children aged 12 years to less than 18 years
200mg on the first day, then 100 mg daily. The length of treatment is dependent on the infection being treated.
Sexually Transmitted Diseases
1 Capsule twice daily for 7 - 10 days.
Primary and Secondary Syphilis
2 Capsules twice daily for 2 weeks. Your doctor will continue to monitor you after your treatment has stopped.
Fevers associated with louse or tick bites
Single dose of one or two Capsules depending on severity.
Treatment of malaria, when chloroquine is not effective 2 Capsules daily for at least 7 days.
Prevention of malaria
1 Capsule daily from 1-2 days before travelling to a malarial area until 4 weeks after returning.
Prevention of scrub typhus
Single dose of 2 Capsules.
Prevention of travellers’ diarrhoea
1 Capsule twice daily on the first day of travel, followed by 1 Capsule daily throughout the stay in the area. If you are planning to take these Capsules for more than 21 days, please consult your doctor.
Prevention of leptospirosis
2 Capsules once each week during the stay in the area; 2 Capsules on completion of the trip. If you are planning to take these Capsules for more than 21 days, please consult your doctor.
You should start to feel better within a few days. If you have been given Respilax for acne it may be a few weeks before you start to see an improvement. If your infection gets worse or you do not start to feel better within a few days (except for acne), or a new infection develops, go back and see your doctor.
Treatment of Rocky Mountain spotted fever
Adults: 100 mg every 12 hours.
Children: weighing less than 45 kg: 2.2 mg/kg body weight given twice a day. Children weighing 45 kg or more should receive the adult dose.
Patients should be treated for at least 3 days after the fever subsides and until there is evidence of clinical improvement. Minimum course of treatment is 5-7 days
If you take more Respilax than you should
If you take too much Respilax contact your doctor or nearest hospital immediately. Always take the labelled medicine package with you, whether there is any Respilax left or not.
If you forget to take Respilax
If you forget to take a Capsule take it as soon as you can. Take your next Capsule at the right time. Do not take a double dose to make up for a forgotten dose.
If you stop taking Respilax
If you stop taking the Capsules too soon, the infection may return. Take the Capsules for the full time of treatment, even when you begin to feel better.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects although not everybody gets them.
Stop taking this medicine and tell your doctor immediately if you experience any of the following serious side effects after taking this medicine. Although they are very rare, the symptoms can be severe.
- Sudden wheeziness, difficulty in breathing, chest pain, fever, swelling of eyelids, face or lips, rash or itching (especially affecting the whole body).
- Upset stomach, loss of appetite, severe, persistent or bloody diarrhoea (this may occur up to two or three months after the last dose and may be associated with stomach pain or fever). This may occur after treatment with antibiotics and can be a sign of serious bowel inflammation.
- Fever, swollen lymph nodes or skin rash. These may be symptoms of a condition known as DRESS (Drug Reaction with Eosinophilia and Systemic Symptoms) and can be severe and life-threatening.
- Very severe headache which may be associated with visual disturbances such as blurred vision, double vision or loss of vision. Permanent visual loss has been reported. The possible symptoms in benign intracranial hypertension include headache, vomiting, visual disturbances including blurred vision, a localized defect in the visual field bordered by an area of normal vision (scotoma), double vision (diplopia), and possible vision loss, in some cases, even permanent.
- Serious illness with widespread severe blistering of the skin, mouth, eyes and genitals.
If any of the side effects listed below occur, contact your doctor as soon as possible.
The Jarisch-Herxheimer reaction which causes fever, chills, headache, muscle pain, and skin rash that is usually self-limiting. This occurs shortly after starting doxycycline treatment for infections with spirochete such as Lyme disease.
Skin that is more sensitive to sunlight than normal. You may get a skin rash, itching, redness or severe sunburn. If this happens stop taking the medicine and tell your doctor.
Inflammation and/or ulcers of the gullet.
Blood disorders.
These are due to changes in numbers of different cell types in the blood.
Symptoms may include tiredness, easy bruising or infections. Low blood pressure.
Increased heart rate. Aches in the joints or muscles.
Stomach pain and diarrhoea.
The side effects listed below may go away during treatment as your body adjusts to the medicine. Tell your doctor if any of these side effects continue to bother you:
Common: may affect up to 1 in 10 people
- feeling or being sick
- worsening of a disease called systemic lupus erythematous (SLE). This is an allergic condition
- which causes joint pain, skin rash and fever headache
- pericarditis (inflammation affecting the heart)
Uncommon: may affect up to 1 in 100 people
- heartburn
- vaginal infection
Rare: may affect up to 1 in 1,000 people
- anxiety
- difficulty in swallowing, sore or painful tongue or mouth
- skin reddening (flushing)
- a ringing or buzzing noise in the ear
- soreness and itching of the rectal and/or genital area
- inflammation of the bowel
- bulging fontanelles (soft spot on head) of infants
- increased pressure in the skull (severe headache with change in vision)
- inflammation and damage to the liver abnormal
- liver function tests
- discolouration of the thyroid tissue when given for long periods. The medicine does not impair thyroid function
- loosening of the nail from the nail bed after exposure to the sun
- increased levels of urea in the blood
- yellow skin and eyes (jaundice), inflammation of the pancreas
- upset stomach, loss of appetite, diarrhoea (this may occur up to two or three months after the last dose), stomach pain
- darker patches on your skin.
Not known: frequency cannot be estimated from the available data
- discolouration and/or lack of growth of teeth
Reporting of side effects
If you get any side effects, talk to your doctor. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine
5. How to store Respilax
Keep this medicine out of the sight and reach of children.
Store below 25ºC.
Capsule should be swallowed whole & not to be opened, chewed or crushed.
Do not use this medicine after the expiry date which is stated on the blister and carton after EXP. The expiry date refers to the last day of that month.
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What Respilax contains
The active substance is doxycycline as Doxycycline Hyclate.
Each hard gelatin capsule contains:
Doxycycline Hyclate IP
equivalent to Doxycycline 100 mg
(as immediate-release pellets)
Lactic acid bacillus 5 billion spores
(as enteric coated pellets)
Colour : Titanium Dioxide IP Excipients q.s.
Approved colours used in the capsule shell.
What Respilax looks like and contents of the pack
Alu strip of 10 capsules.